بیوشیمی در بالین
بیوشیمیبیوشیمی در بالین
بیوشیمیدرباره من
 سلام.من سعی می کنم تست های آزمایشگاهی و کاربرد بالینی و نحوه اندازه گیری آن ها رو در این جا بذارم.امیدوارم به دردتون بخوره.
ادامه...
سلام.من سعی می کنم تست های آزمایشگاهی و کاربرد بالینی و نحوه اندازه گیری آن ها رو در این جا بذارم.امیدوارم به دردتون بخوره.
ادامه...
آمینواسیدوری
![]() مقدمه:
مقدمه:
سال های متمادی از آزمایش ادرار برای غربالگری بیماری های متابولیک استفاده شده است،به خصوص بیماری هایی که ناشی از یک استعداد ژنتیکی بوده اند.در بسیاری از این بیماری ها یک متابولیت غیرطبیعی و یا مقدار بیش از حد یک متابولیت طبیعی در ادرار دفع می شود.
به لحاظ غیر شایع بودن این بیماری ها و غیر اختصاصی بودن علائم آن ها از یک سو و قابل درمان بودن آن ها در صورت تشخیص سریع از سوی دیگر،خون و ادرار را باید با استفاده از تکنیک های بسیار انتخابی و حساس مورد آزمایش قرار داد.
![]() آمینو اسیداوری
آمینو اسیداوری
آمینو اسید در خون از میان غشای گلومرولی فیلتر می شوند ولی به طور نرمال در توبول های کلیوی توسط مکانیسم های انتقال فعال قابل اشباع بازجذب می شوند.
آستانه کلیه برای آمینو اسیدها ی پلاسما بالا است، در نتیجه در حالت طبیعی آمینو اسیدهای بسیار کمی از خون به داخل ادرار فیلتره می شوند. آمینو اسیدها یی که به وسیله ی گلومرول به داخل فیلتره ی گلومرولی وارد می شوند توسط توبول ها مجددا جذب می شوند. آمینو اسیدهایی که در حالت طبیعی به مقدار زیاد (25-200mg/24h) در ادرار دیده می شوند شامل آمینو اسیدهای گلیسین ، تورین ، هیستیدین و گلوتامین می باشند. آمینو اسیدهای دیگر نظیر تریپتوفان ،تیروزین ،سرین ،لوسین ،سیستئین ،آرژنین و فنیل آلانین به مقدار کم (0-25mg/24h) در ادرار افرادسالم وجود دارند.
دفع یک یا تعداد بیشتری از آمینو اسیدها در ادرار می تواند به علت مانعی در مسیر متابولیک (نوع سرریزی) یا کمبود در عملکرد توبولی کلیه (نوع کلیوی) باشد.
![]() تقسیم بندی آمینو اسیداوری روش دنتس
تقسیم بندی آمینو اسیداوری روش دنتس
1. آمینو اسیداوری کلیوی (Renal Aminoaciduria)
آمینو اسیداوری کلیوی معمولا در اثر ضعف(نقص) توبول های کلیوی در بازجذب آمینو اسیدها در ادرار بیمار ظاهر می شود.سطوح پلاسمایی نرمال هستند ولی یک نقص مادرزادی یا اکتسابی در سیستم انتقال کلیوی وجود دارد. آمینو اسیداوری توبولی به 2 دسته تقسیم می شود:
I. آمینو اسیداوری عمومی توبولی (Generalized Tubular Aminoaciduria)که در موارد زیر وجود دارد:در سندرم فانکونی – سیستینوزیس – بیماری ویلسون – گالاکتوزومی
II. آمینو اسیداوری اختصاصی توبولی(Specific Tubular Aminoaciduria) شامل موارد خاص است: سیستین اوری- گلیسین اوری
2. آمینو اسیداوری سر ریز (Overflow Aminoaciduria)
در این جا کلیه هیچ نقصی ندارد .به هر علتی آمینو اسیدهای پلاسما افزایش پیدا کنند که از آستانه ی کلیوی تجاوز می نمایند ، در ادرار بیمار ظاهر می شوند. آمینو اسیداوری سرریز در موارد زیر دیده می شود:
I. عمومی : بیماری های کبدی(چون محل سنتز پروتئین ها در کبد است) – نوزادان نارس (چون هنوز متابولیسم پروتئین ها خوب صورت نمی گیردو به دلیل نقص در متابولیسم پروتئین ها دفع آمینواسیدها در ادرار زیاد است )– آنمی مگالوبلاستیک – مسمومیت با سرب – تحلیل عضلانی– لوسمی
II. اختصاصی : شربت افرا(MSUD) – هارت ناپ – فنیل کتونوری
3.دفع مرضی و غیر طبیعی مشتقات آمینو اسیدها
چنانچه متابولیسم آمینو اسیدها در بدن به روال طبیعی صورت نگیرد، در اثر متابولیسم ناقص ،یک سری موادغیر طبیعی ایجاد می شوند که در ادرار بیمار دفع می شوند.در این حالت خود آمینو اسید در ادرار نیست بلکه مشتقات آن در ادرار ظاهر می شود مثل آلکاپتنوری( که آلکاپتون در ادرار ظاهر می شود)– تیروزینوز -هموسیستینوری
برای پی بردن به نوع آمینو اسید های موجود در ادرار از روش های کروماتوگرافی کاغذی، ستونی یا لایه نازک استفاده می کنند.
![]() آمینو اسید اوری ممکن است اولیه یا ثانویه باشد:
آمینو اسید اوری ممکن است اولیه یا ثانویه باشد:
1. آمینو اسید اوری اولیه
به علت یک نقص آنزیمی ارثی است که اختلال متابولیسم مادرزادی نیز نامیده می شود.نقص در مسیری که یک آمینو اسید خاص متابولیزه می شود یا نقص در سیستم انتقال توبولی خاص کلیوی واقع شده است.
2. آمینو اسیداوری ثانویه
به علت یک بیماری در یک عضو مثل کبد که یک محل فعال متابولیسم آمینو اسید است یا اختلال عملکرد توبولی کلیوی ایجاد شده یا سوءتغذیه انرژی پروتئین
![]() مقادیر طبیعی:
مقادیر طبیعی:
به طور متوسط در بالغین در طول 24 ساعت مقدار 50-200mg آمینو اسید نیتروژن از طریق ادرار دفع می شود.نوزادان به ازاء هر کیلوگرم وزن بدن مقدار 8mg نیتروژن در 24 ساعت دفع می کنند. در نوزادان نارس دفع آمینو اسید های ادرار 4 برابر نوزادان کامل می باشد.درکودکان ، دفع آمینو اسید نیتروژن در 24 ساعت برابر3mg/kg می باشد.
در سیستین اوری( آمینو اسید سیستین) –در فنیل کتونوری(فنیل آلانین)- در بیماری شربت افرا(لوسین و ایزولوسین و والین)-در بیماری هارت ناپ ( گلوتامین و هیستیدین و والین ) در ادرار دیده می شود.
![]() سندرم فانکونی:
سندرم فانکونی:
- به وسیله ی ژن اتوزومال مغلوب به افراد منتقل می شود ، در این بیماری مکانیسم جذب مجدد آمینواسیدها در لوله های ادراری دچار اختلال شده است و از نظر ظهور در فرد به دو صورت دیده می شود:
الف- سندرم فانکونی مادرزادی نوزادان
ب- سندرم فانکونی بالغین
- تشخیص آزمایشگاهی: اگرچه این دو سندرم از لحاظ بالینی با هم متفاوت اند اما از نظر تشخیص آزمایشگاهی نظیر هم می باشند.بیمار مبتلا به گلوکزاوری و پلی اوری با وزن مخصوص ثابت می باشد و دفع پتاسیم و فسفر و اسیداوریک افزایش می یابد در حالی که اسیداوریک خون کاهش می یابد.
- سندرم فانکونی نوزادان اغلب با سیستینوزیس همراه است ؛ در نوع مادرزادی، سیستین در سلول های رتیکولوم مغزاستخوان رسوب می کند که در بیوپسی اندام هایی نظیر کلیه و کبد سیستین را می توان دید.
![]() بیماری ویلسون:
بیماری ویلسون:
- یک نابسامانی متابولیکی و بسیار نادر است که در این بیماری سرولوپلاسمین پلاسما کاهش می یابد (سرولوپلاسمین پروتئینی است که در پلاسما به مس متصل می شود) . بیمار مبتلا به ضعف قوای عضلانی است و حرکاتش کند و بطئی می باشد. حلقه خاکستری مایل به سبز در اطراف چشم بیمار دیده می شود،این حلقه به نام Kayser Fleischer Ring موسوم است.
-یافته های آزمایشگاهی : پایین بودن سطح مس خون – افزایش دفع مس از طریق ادرار – آمینواسیداوری – فسفات اوری، فسفاتمی – کاهش اوریک اسید خون
![]() گالاکتوزومی:
گالاکتوزومی:
- یک بیماری ارثی که توسط ژن اتوزومال مغلوب منتقل می شود.در این بیماری گالاکتوز به گلوکز تبدیل نمی شود. ادرار بیمار حاوی گالاکتوز به مقدار زیاد است و نیز به میزان قابل توجهی دارای آمینواسید می باشد.اگر بیمار تحت درمان سریع قرار نگیرد کبد و مغز او آسیب می بینند.
![]() اختلالات متابولیسم آمینو اسیدها
اختلالات متابولیسم آمینو اسیدها
بسیاری از این بیماری ها نادر و بعید هستند که بیشتر پزشکان با آن مواجه می شوند که اگر درمان نشوند، خیلی از این اختلالات ژنتیکی باعث آسیب مغزی برگشت ناپذیر و مرگ و میر زودرس می شوند.بنابراین تشخیص قبل از تولد یا اوایل پس از تولد و شروع سریع درمان مناسب، در صورت موجود بودن لازم است.
هارت ناپ(Hurtnup)
- یک بیماری ارثی(صفت اتوزومال مغلوب نادر) است که نقص در انتقال(حمل و نقل) روده ای و کلیوی آمینواسیدها ی خنثی از جمله تریپتوفان است که بیشتر (اما نه تمام) شواهد بالینی را می توان به کاهش جذب روده ای و افزایش دفع ادراری تریپتوفان نسبت داد. در این بیماری توبول های کلیوی قادر به جذب مجدد تریپتوفان نیستند. یک آمینواسیداوری عمومی خنثی وجود دارد .
- افزایش دفع مشتقات ایندول که از تخریب تریپتوفان جذب نشده که توسط باکتری های روده به وجود می آیدحاصل می شوند ،از روده جذب شده و در ادرار دفع می شوند.
- به طورطبیعی بخشی از این آمینو اسید به نیکوتین آمید تبدیل می شود،این تبدیل به ویژه اگر مصرف خوراکی نیکوتین آمید در محدوده مرزی باشد ،اهمیت دارد.
- اختلال در جذب روده ای و بازجذب کلیوی تریپتوفان ،در دسترس بودن تریپتوفان را برای بیوسنتز نیاسین محدود می کند و باعث بروز علایم مشابه پلاگر می شود که شامل:
راش قرمز پوسته دار روی نواحی برهنه پوست
آتاکسی مخچه ای برگشت پذیر
اختلال روانی با درجات متغیر
- تشخیص : در این بیماری حداقل 10 نوع آمینو اسید در ادرار بیمار دفع می شود که با روش TLC به خوبی قابل تشخیص می باشد.
1.اختلالات متابولیکی گلیسین
a) گلیسین اوری
توسط ترشح ادراری 0.6-1 gr از گلیسین در روز مشخص می شود که یک گرایشی به تشکیل سنگ های کلیوی اگزالات دارد . گلیسین اوری احتمالاً نتیجه ای از یک نقص در بازجذب توبولی کلیوی است جایی که سطوح گلیسین پلاسمایی نرمال است.
(b هایپراگزالوری اولیه
- در این حالت، ترشح ادراری اگزالات غیر وابسته به جذب رژیم غذایی اگزالات است.
- اگزالات ظاهراً از دآمینه شدن گلیسین افزایش می یابد،با تشکیلGlyoxylate (اگزالات سمی آلدهید)
- نقص متابولیکی شامل یک نارسایی برای کاتابولیزه شدن Glyoxylate است که از این رو به اگزالات اکسید می شود:
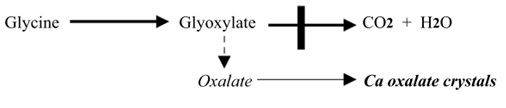
- خصوصیات بالینی شامل سنگ اگزالات کلسیم دو طرفه –عفونت مکرر مجاری ادراری است که با مرگ زودرس از نارسایی کلیوی یا فشار خون دنبال می شود.
- درمان : مصرف پیریدوکسال فسفات (ویتامینB6 ) که یک کو ترانس آمیناز است که ترانس آمیناسیون Glyoxylate را دوباره به گلیسین تحریک می کند.
2.اختلالات متابولیکی آمینواسید های محتوی سولفور
(a Cystinuria (Cystine-Lysinuria)
- یک بیماری متابولیکی ارثی و اتوزومال مغلوب است که با ترشح ادراری سیستین بالای 30 برابر نرمال مشخص می شود که ترشح لیزین ، آرژنین و اورنیتین نیز افزایش می یابد.
- نقص در مکانیسم های بازجذب کلیوی توسط توبول های کلیوی برای این 4 آمینواسید است و وجود سیستین، لیزین ، آرژنین و اورنیتین در ادرار دلیل بر سیستین اوری است و با سیستینوزیس متفاوت است. در این بیماری سطح این 4 آمینواسید در خون بیمار طبیعی یا پایین تر از طبیعی است.
- یک نقص انتقالی مشابه در مخاط روده نشان داده شده است ، اما با وجود کاهش آمینواسیدهای دی بازیک ، به دلیل امکان سنتز آن ها در بدن ، کمبود های آن ها ایجاد نمی شوند.
- از آن جا که سیستین نسبتا نامحلول است،تشکیل سنگ سیستینی در توبول های کلیوی بیماران سیستینوریک رخ می دهد که این نشانه در سیستینوزیس دیده نمی شود.این حالت در افرادهموزیگوت وجود دارد یعنی امکان رسوب و ساختن سنگ در مجرای کلیه وجو دداردولی در هتروزیگوت ها افزایش دفع را می توان نشان داد اما به ندرت در حدی است که ایجاد رسوب کند.
- تشخیص آزمایشگاهی : با روش TLC به راحتی می توان به وجود این 4 آمینواسید در ادرار بیمار پی برد.
- درمان : کاهش دادن غلظت سیستین در ادرار با نوشیدن مقادیر زیادی آب است .قلیایی کردن ادرار، حلالیت سیستین را بالا می برد.اگر این اقدامات ناکافی باشند ، می توان D – پنی سیلامین تجویز کرد ،این دارو با سیستین کمپلکسی می سازد که بسیار محلول تر از سیستین به تنهایی است.
(b سیستینوزیس(بیماری ذخیره سیستین)
- یک اختلال متابولیکی ارثی(اختلال لیزوزومال نادر) است که این وضعیت باید از سیستینوری که یک وضعیت نسبتاً بی خطر است افتراق داده شود.
- نقص در Carrier-mediated Transporter سیستین است.
- کریستال های سیستین در بافت ها و اندام ها خصوصا سیستم رتیکولواندوتلیال رسوب می کنند.با رسوب داخل سلولی کریستال سیستین درون لیزوزوم ها مشخص می شود.کریستال ها ممکن است در کلیه – چشم- مغز استخوان و طحال تجمع کنند.
- در شکل شدید این بیماری،فتوفوبی،نارسایی کلیه ، ریکتز و اختلال رشد دیده می شود.
- با درگیری و آسیب توبول های کلیه به وسیله ی سیستین ، باعث ایجاد سندرم فانکونی می شود و گلوکزاوری و آمینواسیداوری عمومی حادث می شود.
- برخلاف سیستینوری،از دست رفتن سیستین در سیستینوزیس به موازات از دست رفتن سایر آمینواسیدها در ادرار دیده می شود و سیستینوزیز معمولا با آمینواسیداوری عمومی و با منشا کلیوی همراه است. این حالت در سندرم فانکونی دیده می شود که نباید با سیستین اوری اشتباه شود.
- سایر عملکرد های کلیوی نیز به شدت دچار نقص می شوندو بیماران معمولا در جوانی از نارسایی حاد کلیوی می میرند.
(c هموسیستینوریا
- نقص ارثی کاتابولیسم متیونین که توسط ژن مغلوب منتقل می شود.
- در این بیماری متابولیسم متیونین دچار اختلال شده و مقدار زیادی هموسیستین در ادرار بیمار وجود دارد.
نقص به خاطر کمبود سیستاتیونین β سنتاز(CS) است ، ماده ای که تشکیل سیستاتیونین را از هموسیستین و سرین در مسیر متیونین کاتالیز می کند.
- بالای 300میلی گرم هموسیستین ، گاهی با هم با S-آدنوزیل متیونین روزانه در ادرار ترشح می شوند.سطوح پلاسمایی متیونین نیز افزایش می یابد.
- یافته های بالینی شامل عقب ماندگی ذهنی– تشنج-انگشتان عنکبوتی(arachnodactuly)-پوکی استخوان-ترومبوز-تغییرات استخوانی عروقی و تغییراتی که در عدسی چشم بیمار دیده می شود است.
- نمونه ادرار باید تازه باشد زیرا هموسیستین ناپایدار است.
- سیستینوری و هموسیستینوری را می توان با استفاده از روش الکتروفورز با ولتاژ قوی از یکدیگر تشخیص داد.
- درمان : با کاهش متیونین در رژیم غذایی و تجویز پیریدوکسین است.
3.اختلالات متابولیکی فنیل آلانین و تیروزین
(a فنیل کتونوری (PKU)یا فنیل آلانینمیا
- یک بیماری ارثی اتوزومال مغلوب است.اختلالات عمده متابولیکی در ارتباط با اختلال توانایی برای تبدیل فنیل آلانین به تیروزین است.
- فنیل آلانینمی : فنیل آلانین یکی از آمینواسیدهای ضروری بدن می باشد که از متابولیسم این آمینواسید مواد مختلفی تولید می شود. فنیل آلانینمی زمانی اتفاق می افتد که در یکی از آنزیم هایی که در متابولیسم این آمینواسید دخالت دارند کمبود یا فقدان وجود داشته باشد.فنیل آلانینمی با PKU همراه است.

- نقص در این مسیر تبدیلی ممکن است به علل زیر باشد:
1.فقدان یا کمبود فنیل آلانین هیدروکسیلاز(PKU کلاسیک یا نوع I)
2.نقص در دی هیدروبیوپترین ردوکتاز (نوع II و III)
3. نقص در دی هیدروبیوپترین بیوسنتاز(نوع IV وV )
- مهم ترین پیامد PKU نوع I درمان نشده ، عقب ماندگی ذهنی،تشنج ، اگزما و بوی موش است.
- سطوح پلاسمایی فنیل آلانین و فنیل پیروییک اسید و سطح ادراری فنیل پیروییک اسید(بالاترین) ، فنیل استیک اسید و فنیل آلانین افزایش نشان می دهند.
- یافته های آزمایشگاهی شامل : افزایش فنیل آلانین و متابولیت های فنیل آلانین (فنیل پیروییک اسید- فنیل لاکتیک اسید – فنیل استیک اسید ) در خون و ادرار است.
- به عنوان علامت مشخصه ادرار و عرق این بیماران به علت فنیل استیک اسید، دارای بوی موش یا کپک می باشد.
-تیروزین خون بعد از Loud فنیل الانین افزایش نمی یابد.
- درمان : با محدودیت فنیل آلانین در رژیم غذایی

تیروزینمیا(تیروزینوز)-تیروزینوری
- یک نارسایی متابولیسمی و یک بیماری ارثی است.
- تیروزینمی همراه با تیروزینوری زمانی اتفاق می افتد که تیروزین مشتق از رژیم غذایی یا فنیل آلانین دارای متابولیسم غیر طبیعی باشد که ممکن است قسمتی از یک اختلال عمومی آمینواسیدها همراه با بیماری کبدی یا نشان دهنده ی یکی از اختلالات ژنتیکی متابولیسم تیروزین باشد.
- نقص : تیروزینمی ارثی نوعI(تیروزینوز) : کمبود فوماریل استواستات هیدرولاز(FAA Hydrolase)
تیروزینمی نوعII : کمبود تیروزین ترانس آمیناز
- یافته های آزمایشگاهی شامل : افزایش تیروزین و متابولیت های آن پارا- هیدروکسی فنیل پیروییک اسید ،استیک و لاکتیک اسیدها
- یافته های بالینی : سیروز کبدی،انبساط شکمی ،بزرگی طحال ، آسیب کلیوی که ممکن است باعث مسمومیت آمونیا(ammonia) و باعث گلوکزاوری کلیوی شود.
- درمان : محدود کردن رژیم غذایی فنیل آلانین – تیروزین –متیونین
1.فقدان یا کمبود فنیل آلانین هیدروکسیلاز(PKU کلاسیک یا نوع I)
2.نقص در دی هیدروبیوپترین ردوکتاز (نوع II و III)
3. نقص در دی هیدروبیوپترین بیوسنتاز(نوع IV وV )
- مهم ترین پیامد PKU نوع I درمان نشده ، عقب ماندگی ذهنی،تشنج ، اگزما و بوی موش است.
- سطوح پلاسمایی فنیل آلانین و فنیل پیروییک اسید و سطح ادراری فنیل پیروییک اسید(بالاترین) ، فنیل استیک اسید و فنیل آلانین افزایش نشان می دهند.
- یافته های آزمایشگاهی شامل : افزایش فنیل آلانین و متابولیت های فنیل آلانین (فنیل پیروییک اسید- فنیل لاکتیک اسید – فنیل استیک اسید ) در خون و ادرار است.
- به عنوان علامت مشخصه ادرار و عرق این بیماران به علت فنیل استیک اسید، دارای بوی موش یا کپک می باشد.
-تیروزین خون بعد از Loud فنیل الانین افزایش نمی یابد.
- درمان : با محدودیت فنیل آلانین در رژیم غذایی

آلکاپتنوری
- یک اختلال متابولیکی ارثی است که توسط ژن مغلوب به ارث می رسد.
- به طور طبیعی فنیل آلانین و تیروزین به هموژنتیسیک اسید (دی هیدروکسی فنیل استیک اسید) متابولیزه می شوند، که آن هم به مالئیل استو استیک اسید اکسید می شود.
- نقص به دلیل فقدان هموژنتیسیک اسید اکسیداز کبدی است که افزایش هموژنتیسیک اسید که به طور طبیعی از متابولیسم فنیل آلانین ایجاد می شود که دربافت ها و خون تجمع می یابد، وجود دارد که در ادرار می ریزد.
- اکسیداسیون و پلی مریزاسیون هموژنتیسیک اسید باعث تولید پیگمان آلکاپتون می شود که مسیر آن شبیه پلی مریزاسیون DOPA برای تولید ملانین است.
- بیمار در سنین جوانی ناراحتی به خصوصی ندارد ولی با بالا رفتن سن ، پیگمان های سیاهی در استخوان و مفاصل ظاهر شده و بیماری خاصی به نام اکرونوزیس به وجود می آید که در اثر رسوب آلکاپتون در غضروف ها و تیرگی حاصل از ان است و سبب تیرگی غضروف های گوش و معمولاً آرتریت در سنین بالا می شود.
- یافته های بالینی : آرتریت(التهاب مفاصل) دژنراتیو – پیگمانتاسیون غضروف
- تشخیص : با آزمایش هموژنتیسیک اسید در ادرار- روش های تاییدی شامل کروماتوگرافی کاغذی یا لایه نازک و الکتروفورز موئینگی است.این روش ها باید هموژنتیسیک اسید را از ژانتیزیک اسید ( یک متابولیت آسپرین) افتراق دهند.
- ادراری که حاوی این اسید باشد ظاهری معمولی دارد ولی در اثر ماندن یا قلیایی شدن به رنگ قهوه ای یا سیاه در می آید.تبدیل هموژنتیسیک اسید به آلکاپتون در شرایط قلیایی تسریع می شود و مشخص ترین ناهنجاری در آلکاپتنوری ،تیرگی ادرار است که با راکد ماندن قلیایی تر شده است،اما این یافته همیشه وجود ندارد.در صورت وجود ، اولین بار مادر بچه از این وضعیت نگران می شود به این صورت که کهنه ی بچه سیاه است و با شستشو در شوینده های قلیایی سیاه تر می شود.
- درمان : در دسترس نیست.
4.اختلالات متابولیکی متابولیسم آمینواسیدهای شاخه دار(والین – لوسین – ایزولوسین)
بیماری شربت افرا MSUD (Maple Syrup Urine Disease): Branched Chain Ketonuria
- یک اختلال مغلوب اتوزومال ارثی است که مرتبط با متابولیسم غیرطبیعی زنجیره جانبی آمینواسیدها است.
- نقص بیوشیمیایی فقدان یا کاهش شدید فعالیت α-کتواسید دکربوکسیلاز است.

- در این بیماری آمینواسید های شاخه دار مانند والین – لوسین – ایزولوسین متابولیزه نمی شوند و این آمینواسیدها به صورت α-کتواسید مربوطه اشان در خون تجمع یافته و در ادرار بیمار ظاهر می شوند.
- سطوح پلاسمایی و ادراری والین – لوسین – ایزولوسین و α-کتواسیدها افزایش می یابد.
- به علت شباهت بوی ادرار با بوی شربت افرا این نام را برای بیماری به کار می برند یا قند سوخته یا مواد قندی حامل کارامل یا زردچوبه هندی را دارد.در هر صورت علت آن نامشخص است.
- بچه به سختی غذا می خورد و ممکن است استفراغ کند و نیز تشنج ، گیجی ، تنفس های نامنظم و اغلب هیپوگلیسمی مشخص می شود.
- آسیب مغزی شدید در کودکان بازمانده رخ می دهد.بدون درمان مرگ معمولا تا پایان یک سال رخ می دهد.
- تشخیص قبل از یک هفتگی فقط توسط آنالیز آنزیمی یا ژنتیکی صورت گیردو این بیماری در هفته اول زندگی ظاهر می شودو اگر درمان صورت نگیرد ، ضایعات نورولوژیک شدید ایجاد شده و سبب مرگ در چند هفته یا ماه اول خواهد شد.
- تشخیص : نشان دادن افزایش غلظت آمینو اسیدهای منشعب در پلاسما و ادرار
- درمان در هفته اول زندگی تا حد زیادی از عواقب جلوگیری می کند.
درمان : جایگزینی پروتئین غذایی با یک مخلوطی از امینو اسید ها که شامل مقادیر اندکی از آمینواسید های با زنجیره منشعب والین – لوسین – ایزولوسین است ،تکامل نرمال محتمل خواهد بود.
وجود مخاط در ادرار mucus in urine
وجود مخاط در ادرار mucus in urine
مقدمه :
عفونت ادراری یکی از شایع ترین عوارض یوِیژه در بین خانومهاست چون طول آلت تناسلی آن کوتاه است و فاصله کمی با مقعد دارد و شستن باید از جلو به عقب باشد و نه به عکس تا باکتریهایی که در مدفوع و روده بزرگ هستند وارد واژن و مجاری ادرار زنها نشود چون کافی است خانوم دچار عفونت ادراری شود و همین عفونت را بسادگی در طی مقاربت به شوهرش هم منتقل کند واین آغاز داستان و گرفتاری های طولانی است .
در اینجا به بحث تخصصی در باره یکی از مشکلات که وجود موکوس = مخاط = mucus در ادرار است پرداخته شده است این یکی از فاکتورهایی است که در آزمایش ادرار اندازه گیری می شود و آنالیز آن می تواند راهگشا باشد از مشکلات ساده مثل عفونت ادراری تا حتی تومورهای بدخیم و سرطان و راه خوبی برای تشخیص به موقع و پیشگیری از برخی بیمارهاست .
در مناطق گرمسیر جنوب و مرکز ایران بدلیل گرمی و خشکی هوا و از دست رفتن آب بدن و از همه بدتر ننوشیدن آب کافی و سالم ، بدن با کمبود آب مواجه می شود و به اصطلاح دهیدراته شده ادرار غلیظ می گردد و بدن مساعد انواع بیماریها از جمله سنگ کلیه و همین عفونت مجاری ادراری می شود . به همین جهت حتی اگر تشنه نیستید باید هر روز مقدار زیادی آب بخورید چون اولا با بالا رفتن سن سیگنالهایی که احساس تشنگی را در ما ایجاد می کنند ضعیف می شوند و در واقع با وجودیکه تشنه هستیم این تشنگی را حس نمی کنیم و از آن گذشته وقتی احساس تشنگی می کنید که بدن با کمبود آب مواجه شده و کمی دیر شده است.
موکوس در ادرار
هر گاه در ادرار موکوس وجود داشته باشد این مشکل چندین علت می تواند داشته باشد در اغلب موارد بیمار دچار یوتی آی " TUI=urinary tract infection = عفونت مجاری ادراری " می باشد که منحصراً مثانه را درگیر کرده است و یا بر روی کلیه ها هم اثر گذاشته است . موکوس = مخاط در ادرار معمولاً زرد رنگ و بصورت کلوئیدی چسبناک viscous colloid می باشد .
موکوس یک پروتئین فیبری است که اغلب در رسوب ادرار افراد دیده می شود .رشته های موکوسی به صورت موجی و گاهی به صورت خط مستقیم دیده می شوند.رشته های موکوسی ممکن است با سیلندر هیالین اشتباه تشخیص داده شوند ولی شکل استوانه ای سیلندر ها و انتهای گرد آن ها ، سبب تفکیک آن ها از رشته های موکوسی می شود.
مجاری ادراری تناسلی به خصوص اپی تلیوم واژن منشا بیشترین میزان موکوس موجود در ادرار خانم ها است.
وجود مخاط در ادرار باعث می شود که رنگ آن کدر شود در حالیکه ادرار طبیعی شفاف است.
وجود مخاط در ادرار نشان دهنده این است که مشکلی وجود دارد
وجود مخاط درادار مشکلی است که باید حل شود باید منشاء آن پیدا شود و مداوا گردد . چون مخاط در ادرار یافته ای طبیعی نیست و عملا در ارتباط با برخی مشکلات دیگر است . چندین علت احتمالی برای این مشکل وجود دارد که برخی از آنها در اینجا فهرست شده است :
1- عفونت و التهاب مجاری ادراری به خصوص تورم مزمن پیشابرا ه و مثانه : همانگونه که قبلا هم ذکر شد یکی از علل آن می تواند عفونت مجاری ادراری UTI باشد که این عمده ترین دلیل است .
2- مشکل می تواند در ارتباط نزدیک با برخی بیماری های مقاربتی Sexually transmitted disease = STD باشد بویژه آنهایی باشد که جدا از مجاری تناسلی باعث آلودگی مجاری ادراری هم می شوند .
3- سنگ کلیه : تحت شرایط ویژه این مشکل می تواند بدلیل سنگ کلیه هم باشد ارتباط بین سنگ کلیه و موکوس می تواند بشکل ثانویه باشد یعنی سنگ می تواند باعث بروز عفونت در مجاری ادراری شود.
4- تومورهای بدخیم : یکی از علت های وجود مخاط در ادرار می تواند تومور بدخیم = سرطان در مجاری ادرار باشد در این حالت رنگ ادرارتغییر می کند و این بدلیل وجود مخاط است .
یافته های بیشتر
تجزیه و تحلیل ادرار کمک عمده ای به تشخیص عفونت مجاری ادراری می کند . بررسی ظاهر ادرار و وجود موکوس در آن بو و مشخصه های آن به همراه نشانه ها و شکایت هایی که بیمار دارد باید با هم مد نظر گرفته شوند . این آزمایش بطور معمول افزایش گلبولهای سفید در ادرار white blood cell in urine را نشان می دهد ( عاملی که نشان دهنده پیشرفت عفونت است )
توجه کنید که زنان وقتی می خواهند نمونه ادرار بگیرند باید توجه بیشتری بخرج دهند چون مجرای تناسلی می تواند در مجاورت ترشحات واژن قرار گرفته و با ادرار مخلوط شده و سبب عفونت ادراری شود .
منابع:
ادرار و مایعات بدن-دیوید سون
http://ppn.persianblog.ir/tag/mucus_in_urine
نام آزمایش : اندازه گیری پروتئین در مایع مغزی نخاعی یا پونکسیون کمری Lumbar puncture(LP) به روش توربیدومتری(کدورت سنجی)
بسمه تعالی
نام آزمایش : اندازه گیری پروتئین در مایع مغزی نخاعیCerebrospinal Fluid(CSF) یا پونکسیون کمری Lumbar puncture(LP) به روش توربیدومتری(کدورت سنجی)
![]() مقدمه :
مقدمه :
مایع مغزی نخاعی که مایع نخاعی نیز نامیده می شود، در بطن های مغز و در میان طناب نخاعی جریان دارد و به طور کامل مغز و طناب نخاعی را در بر می گیرد و در نتیجه این ساختمان ها را در مقابل صدمات حمایت و محافظت می کند و نیز مانند لنف فرآورده های زائد متابولیسم را حذف می کند. مایع مغزی نخاعی بسیار به آهستگی گردش می کند ، لذا زمان تماس با سلول های سیستم عصبی مرکزی افزایش می یابد.از150-200 میلی لیتر CSF ، تقریبا 100 میلی لیتر به وسیله ی خون در بطن های مغز تولید شده و روزانه به داخل جریان خون بازجذب می شود. مایع مغزی نخاعی عمدتاً به وسیله ی اولترافیلتراسیون پلاسما از دیواره های مویرگی شبکه کوروئید(دسته ای از عروق خونی کوچک) در بطن های جانبی مغز تشکیل می شود. این شبکه همچنین مقادیر جزئی از مواد مانند کلراید را ترشح می کند.
تجزیه و تحلیل مایع نخاع معمولاً شامل رنگ ، فشار ، شمارش سلول (WBC یا RBC خون ) ، پروتئین ، کلراید و گلوکز می باشد.سطوح پروتئین و گلوکز CSF از سطوح خونی پائین تر هستند ، با این حال ، سطوح کلراید CSF بالاتر از سطح کلراید سرم می باشد. CSFازپالایش پلاسما تشکیل می شود و سومین مایع اصلی بدن است.یک سیستم فیزیولوژیکی جهت تأمین مواد غذایی برای بافت عصبی ، حرکت مواد زاید حاصل از سوخت وساز و ایجاد یک سد مکانیکی برای حمایت مغز و طناب نخاعی از ضربه و نرم سازی بافت عصبی مهیا می سازد. مقادیر مرجع به ترتیب زیر است:
|
|
رنگ |
شمارش سلول (لکوسیت ها) میکرولیتر |
پروتئین(mg/dl) |
کلراید(mEq/dl) |
گلوکز(mg/dl) |
|
بالغین |
بی رنگ و شفاف |
0-8 |
15-45 |
118-132 |
40-80 |
|
کودکان |
بی رنگ و شفاف |
0-8 |
15-45 |
120-128 |
35-75 |
|
نوزادان نارس |
|
0-20 |
کمتر از 400 |
|
|
|
نوزادان |
شفاف |
0-15 |
30-200 |
110-122 |
20-40 |
|
1-6ماهه |
|
|
30-100 |
|
|
1. ظاهر : مایع نخاع طبیعی شفاف و بیرنگ بوده و میزان چسبندگی آن مانند آب است.
2. بررسی رنگ :
I. گزانتوکرومیXanthochromia
مایع نخاع را سانتریفوژ نموده ، مایع رویی را بررسی می کنیم ، چنانچه به رنگ صورتی ، نارنجی یا زرد باشد آن را گزانتوکروم می نامیم.رنگ زرد نشاندهنده ی خون کهنه (4 الی 5 روز بعد از خونریزی مغزی) است ، مخلوط شدن بیلی روبین وخون ، سطوح پروتئینی بیش از حد بالا ، و تغییر رنگ مایع به طور طبیعی به مدت 3 هفته باقی می ماند.
علل گزانتوکرومی :
1. ماندن مایع نخاع خون آلود در خارج یخچال به مدت بیش از یک ساعت : هموگلوبین تغییر یافته
2. یرقان : وجود بیلی روبین می تواند در زرد شدن CSF نقش داشته باشد.
3. بالا بودن پروتئین مایع نخاع بیش از 150 میلی گرم در دسی لیتر : غلظت بسیار بالای پروتئین مثلاً به دلیل تومورهای مغزی که گردش CSF را مختل می کند.نمونه های این بیماران به دلیل وجود فیبرینوژن ، بلافاصله لخته می شوند.
4. رنگدانه های کاروتینوئید، ملانین و ریفامپین
II. ظاهر ابری
ازدیاد گلبول های سفید (چرک) : معمولاً از روی کدورت شدید نمونه مشخص است.وجود سلول های چرکی به وسیله ی میکروسکوپ قابل شناسایی است. ظاهر ابری ممکن است نشان دهنده ی افزایش شمارش WBC یا سطح پروتئین باشد.
III. رنگ قرمز روشن
ü تعیین منشأ خون آلود بودن مایع نخاع :
1. در خونریزی ناشی از جراحت نمونه برداری به تدریج در لوله های دوم و سوم خون کمتر می شود ونیز در پونکسیون تروماتیک خون CSF لخته خواهد شد.
2. گزانتوکرومی ، مشاهده میکروسکوپی اریتروفاگوسیتوز و ماکروفاژهای حاوی هموسیدرین نشانگر خونریزی هستند به شرط آن که قبلا جراحت ناشی از نمونه گیری وارد نشده باشد.بعد از جراحت نمونه گیری 1 تا 2 ساعت طول می کشد تا گلبول های قرمز لیز شوند و لذا باید سریعاً نمونه آزمایش شود.در هموراژی زیر عنکبوتیه لخته به وجود نمی آید.
اگر مایع نخاعی به صورت 3 نمونه مجزا گرفته شده باشد ، در حالت خونریزی جدید در فضای زیر عنکبوتیه هر 3 نمونه به طور یکسان به رنگ خون در می آیند ولی در آسیب عروق خونی طی پونکسیون کمری شدت رنگ به طور پیشرونده ای کاهش می یابد . وجود گلبول های قرمز در مایع نخاع رنگ آن را به سمت صورتی یا قرمز سوق می دهد.چنانچه تعداد گلبول های قرمز از 6000 عدد در میکرولیتر بیشتر باشد ، نمای مایع نخاع کاملاً خون آلود جلوه می کند.
3. کدورت
ü ایجاد کدورت در Csf می تواند به دلایل ذیل باشد:
1. معمولاً به دلیل چرک است.
2. تعدادلکوسیت بیش از 200 عدد در میکرولیتر
3. تعداد گلبول قرمز بیش از 400 عدد در میکرولیتر
4. وجود میکروارگانیسم ها (باکتری ، قارچ ، آمیب)
5. مواد رادیو گرافیک
6. افزایش پروتئین (مثل مننژیت باکتریایی ، ویروسی ، قارچی و...)
جهت بررسی ظاهر مایع نخاع آن را در برابر نور مستقیم خورشید گرفته با زاویه 90 درجه به آن نگاه کنید . وجود بیش از 50 سلول در میکرولیتر نمایی شبیه به دانه های برف یا ذرات براق در مایع نخاع ایجاد می کند.
4. لخته شدن سریع
در موارد ذیل ممکن است در مایع نخاع لخته مشاهده شود :
1. جراحت ناشی از نمونه گیری Traumatic tap
2. انسدادکامل نخاع(سندرم فریونFroin)
3. مننژیت چرکی و یا سلی(توبرکلوزی)
4. وقتی مقادیر زیادی فیبرینوژن در نمونه وجود داشته باشد( که معمولاً با غلظت بسیار بالای پروتئین همراه است) ، لخته ایجاد می شود.
5. تومورهای CNS
5. ارزیابی های بیوشیمیایی : گلوکز – پروتئین –لاکتات دهیدروژناز – اسیدلاکتیک – گلوتامین -کلراید
![]() پروتئین
پروتئین
به دلیل این که پروتئین مولکول بزرگی است و از سد خونی مغزی عبورنمی کند مقدار طبیعی آن در CSF بسیار کم است.به دلیل این که آلبومین کوچک تر از گلوبولین است و می تواند راحت تر از سد خونی مغزی عبور کند ، نسبت آلبومین به گلوبولین در CSF به طور طبیعی بیشتر از پلاسمای خون می باشد. مقدار طبیعی پروتئین مایع نخاع بین 15-45 mg/dl است که در نوزادان تا 150mg/dl و در نوزادان نارس تا 500mg/dl هم مشاهده می شود.در نوزادان به دلیل نفوذ پذیری نسبتاً بالای عروق ، غلظت پروتئین CSF حدود 3 برابر بزرگسالان است.تعیین پروتئین ، فراوان ترین آزمون شیمیایی است که روی CSF انجام می شود.
بیماری مغزی می تواند به دو دلیل غلظت توتال پروتئین و نسبت اجزای آن را در CSF تغییر دهد.1) افزایش نفوذ پذیری عروقی و مننژی می تواند سبب ورود پروتئین بیشتر به CSF شود. 2) ممکن است سلول های التهابی یا سایر سلول های مهاجم ، در کانال مغزی نخاعی پروتئین (ایمنوگلوبولین) سنتز کنند.(افزایش سوخت و ساز سلول های عصبی در پاسخ به حالات پاتولوژیک)
![]() موارد افزایش پروتئین در مایع نخاع :
موارد افزایش پروتئین در مایع نخاع :
1. پونکسیون تروماتیک نخاعی
2. افزایش نفوذ پذیری خونی-Csf :
I. مننژیت(باکتریایی، ویروسی ، سلی و...)
II. خونریزی یا هموراژی (زیر عنکبوتیه ، داخل مغزی)
III. اختلالات اندوکرین/متابولیک مانند کاهش عملکرد اندوکرین ( تیروئید ، پاراتیروئید)
IV. مسمومیت داروئی (اتانول ، فنی توئین )
3. نقایص جریان csf
I. انسدادمکانیکی (تومور مغزی ، آبسه)
II. تجمعات حفره ای Csf
4. افزایش ساخت IgG
I. نوروسفلیس
5. افزایش ساخت IgG
6. و با نفوذ پذیری خونی-Csf
I. سندرم گیلن باره
![]() موارد کاهش پروتئین در مایع نخاع:
موارد کاهش پروتئین در مایع نخاع:
1. برداشت مقدار زیادی از مایع نخاع
2. نشت مایع نخاع بر اثر ضربه یا نمونه برداری
3. افزایش فشار داخل جمجمه
4. هیپرتیروئیدیسم
اندازه گیری پروتئین توتال CSF یک تست نسبتاً غیر حساس برای تشخیص بیماری مغزی است ، زیرا تغییرات غلظت یک پروتئین خاص ، همیشه باعث افزایش قابل کشف در غلظت توتال پروتئین نمی شود.اندازه گیری های سریال را می توان جهت مانیتور کردن درمان به کار برد.
6. ازریابی میکروسکوپی (هماتولوژیک) : سیتولوژی : شمارش کلی و افتراقی سلول ها برای کمک به تشخیص بیماری ها
7. ارزیابی میکروبیولوژیک : رنگ آمیزی لام گرفته شده از CSF و بررسی میکروسکوپی آن و نیز کشت آن در بررسی مننژیت باکتریایی – نوروسفلیس – مننژیت قارچی و...
8. ارزیابی سرولوژیک:آزمون های سرولوژیکی بررسی بیماری های التهابی مانندCRP ویا تست VDRL برای بیماری سفلیس
شیوه انجام تست :
پس از فراهم کردن لوازم زیر : یک سینی استریل سوراخ کردن کمر ، یک محلول گند زدا(پوویدون آیوداین یا ید ) ، یک ماده ی بی حسی موضعی ( مثلا لیدوکائین) ، یک جفت دستکش استریل و نوار چسب ، بیمار را در وضعیت جنینی قرار داده ، با پشت(کمر) خمیده ، سر خم شده روی سینه و زانو های کشیده شده به طرف شکم قرار دهید.معمولاً مایع نخاعی به وسیله ی پونکسیون لومبار جمع آوری می شود.به بیمار بگوئید دستان خود را دور زانوان قلاب کند تا بتواند این حالت را حفظ کند.می توان از وضعیت نشسته هم استفاده کرد.
روی سه لوله آزمایش برچسب 1و2و3 بزنید.پزشک فشار مایع نخاعی را با استفاده از یک مانومتر متصل به سوزن بررسی می کند .پزشک جمعاً 10 تا 12 میلی لیتر ( یا 6 میلی لیتر مایع نخاعی به صورت اجزاء 2 میلی لیتری)مایع نخاعی جمع آوری می کند : 3 میلی لیتر در لوله شماره 1 ،3 میلی لیتر در لوله شماره 2 و 3 میلی لیتر در لوله شماره 3 می ریزد.اولین لوله می تواند آلوده شده باشد ( با خون ناشی از کشیدن مایع نخاع) ونباید برای شمارش سلول ، کشت یا سنجش پروتئین مورد استفاده قرار می گیرد. مایع نخاعی را به وسیله ی سوراخ نمودن کمر (Spinal tap) که در کیسه ی کمری بین مهره ی کمری 3-4 L یا بین مهره ی کمری 4-5 L انجام می شود به دست می آورند.در ابتدا فشار CSF را اندازه گیری می کنند.سپس مایع را آسپیره کرده و در لوله های آزمایش استریل می ریزند.ابتدا برای آزمایشات میکروبیولوژیک فرستاده می شود و در صورت لزوم بقیه نمونه برای آنالیز شیمیایی قابل استفاده است.اگر تست ها به ترتیب عکس انجام شوند احتمال آلودگی باکتریال نمونه وجود دارد.
لوله شماره 1 برای آزمایش های بیوشیمیایی و سرولوژیکی ، لوله شماره 2 برای میکروب شناسی و لوله شماره 3 برای شمارش سلولی به کار می رود زیرا احتمال کمی وجود دارد که سلول های حصل از پونکسیون مایع نخاعی وارد لوله شماره 3 شوند.
روش های اندازه گیری پروتئین CSF :
از روش های متداول اندازه گیری پروتئین ، می توان به توربیدومتری - کالریمتری اشاره کرد.
برای اندازه گیری فراکسیون های پروتئین CSF از قبیل آلبومین و گلوبولین الکتروفورز و ایمنودیفیوژن شعاعی(RID) و نفلومتری استفاده می شود.
1. روش های کدورت سنجی :
استفاده از معرف های اسید سولفوسالیسیلیک و تری کلرواستیک اسید و سولفات سدیم برای رسوب پروتئین در اندازه گیری پروتئین مایع نخاع معمول است که هر دو دارای محدودیت هایی می باشند.سولفوسالیسیلیک اسید 1.5 % باعث دناتوره کردن پروتئین های Csf و کدورت نمونه می شود که به وسیله ی اسپکتروفوتومتری قابل اندازه گیری است.
برای اندازه گیری مقدار آن روش بیوره دقیق نیست ، جون مقدار پروتئین خیلی کم است و روش فولین سیوکالتو هم مناسب نیست چون مقدار آمینواسید های Csf به نسبت برای این روش زیاد بوده و جواب آزمایش دقیق به دست نمی آید.از طرف دیگر افزایش پروتئین Csf در اغلب بیماری ها با افزایش دو جزء مهم پروتئین یعنی آلبومین و گلوبین است ، لذا در بیشتر آزمایشات مقدار گلوبولین را اندازه گیری می کنند به جای پروتئین تام Csf .
2. روش های رنگ سنجی :
شامل روش لوری ، روش های متصل به رنگ ( که از کوماسی بریلیانت بلو یا پانسوs استفاده می کنند ) ، و روش بیوره تعدیل یافته ، می باشد.روش کوماسی بریلیانت بلو نسبتاً شایع است زیرا سریع و خیلی حساس بوده و با مقادیر کم نمونه قابل انجام است.
روش آزمایش : اندازه گیری پروتئین تام مایع نخاع : طریقه ی کدورتی
0.5ml Csf را با 4/5 میلی لیتر اسید سولفوسالیسیلیک 1/5 درصد مخلوط نموده ، به مدت 5 دقیقه در دمای اتاق انکوبه نمائید و در مقابل بلانک آب مقطر در طول موج 525 نانومتر بخوانید.
محاسبه در طول موج 525 نانومتر :
100 * 1.7 * OD = مقدار پروتئین بر اساس mg/dl
سیستاتین c و نقش آن در تشخیص و پیگیری در بیماران پیوند کلیه
سیستاتین c و نقش آن در تشخیص و پیگیری در بیماران پیوند کلیه
1.مقدمه
پروتئازها آنزیم هایی هستند که هیدرولیز باندهای پپتیدی در پروتئین ها و پپتید ها را کاتالیز می کنند. براساس گروه های شیمیایی مسئول برای کاتالیز ،پروتئازها به5 گروه اصلی طبقه بندی می شوند:سرین ،سیستئین، آسپارتیک ،ترئونین و متالوپپتیدازها.
فعالیت آن ها به شدت در سطوح مختلفی تنظیم می شود.پروتئولیز کنترل نشده منجر به آسیب به ماشین بیولوژیک سلول ها و بافت ها می شود و منجر به در جات پاتوفیزیولوژیک که سرانجام باعث مرگ ارگانیسم می شود.
مهار پروتئاز ها توسط مهار کننده های پروتئینی مثل سیستاتین هاصورت می گیرد که یکی از مهم ترین مکانیسم های تنظیم فعالیت پروتئازها را نشان می دهد.
سیستاتین ها یک ارگانیسم را از فعالیت مضر پروتئازهای اندوژن هدف و نیز از پروتئازهایی که از ویروس ها-باکتری ها و پروتوزوآهای عفونی آزاد می شود محافظت می کند.
2.سیستاتین ها
سیستاتین ها مهار کننده های پرو تئینی سیستئین پرو تئازهای خانواده ی پاپائین هستند.
سیستاتین ها پروتئازهای هدف را به طور برگشت پذیر و رقابتی توسط مسدود کردن غیر مستقیم مراکز کاتالتیک این آنزیم ها غیر فعال می کنند
بنابر این مانع برش سوبسترامیشوند.آن ها تشکیل کمپلکس بیومولکولار پایداری با پرو تئاز ها می دهند که این غیر فعال شدن را ساعت ها و هفته ها نگه می دارند.
ابر خانواده ی سیستاتین متشکل از پروتئین هایی با ساختمان ها اولیه اندازه ها و توزیع های گوناگون است،پروتئین هایی با یک یا چند دومن ،بایا بدون باندهای دی سولفیدی و نیز قندی یا غیر قندی اند.
توالی های آمینو اسیدی آن ها پیشنهاد می کند که سیستاتین ها از یک اجدادپروتئینی مشترک بدون باند دی سولفیدی منشا می گیرن و آن یک پروکورسور محتوی باند ها در حدود 1000میلیون سال پیش است.
پروتئین های ابر خانواده ی سیستاتین قبلا بر اساس تشابهات توالی آن ها به سه خانواده ی اصلی تقسیم می شوند.ویژگی های اصلی برای طبقه بندی ستیاتین ها اندازهای زنجیرها پلی پپتید و حضور یا غیاب باندهای دی سولفیدی داخلی شد.
3.اعمال فیزیولوژیک سیستاتین ها
1.نقش محافظتی
-در بیماری های التهابی و خود ایمنی
-مهار پروتئاز های اگزوژن
سیستاتین ها به طور گسترده ای در حیوانات، گیاهان و پروتوزوآ توزیع شدند به صورت داخل سلولی و خارج سلولی که اشاره به نقش فیزیولوژیکی مهم آن ها دارد.آن ها معتقدند که سلول ها و ارگانیسم ها را از فعالیت کنترل نشده ی سیستئین پرو تئازهای اندوژن آزاد شده از لیزوزوم های سلول های در حال مرگ و مرده محافظت می کند و نیز مهارکننده های مهم سیستئین پروتئازها توسط میکروارگانیسم ها و پارازایت ها برای هجوم سلول های میزبان و داخل شدن ارگانیسم ها استفاده شده است.
2.نقش های تنظیمی
-تمایز
-تنظیم پرولیفراسیون سلولی
4.سیستاتین c
نام دیگر سیستاتین c ، -Trace Ɣ یا -globulinƔ post یا gamma csf است.
یک مهار کننده ی سیستئین پروتئاز های مختلف در جریان خون است..وزن مولکولی13,343Da است که توسط اسپکترومتری جرمی و محاسبه از طریق توالی اسید آمینه ای تک زنجیره ی پلی پپتیدی آن به دست آمده است.در حدود 50% از سیستاتین c یک پرولین هیدروکسیله شده در سه موقعیت را محل می کند و وزن مولکولی این هیدروکسیله شده13359 دالتون می شود. ساختار کریستالی سیستاتین c شناخته شده است.
نقطه ی ایزو الکتریک سیستاتین c 9.3 است و بنابر این در همه ی مایعات بدن بار + دارند.
سیستاتین c فراوان ترین مهار کننده این ابر خانواده ی سیستاتین در همه ی مایعات بدن به خصوص در بالاترین غلظت در پلاسمای سمینال و مایع مغزی نخاعی است.
توزیع فراوان این پروتئین به نقش ضروری آن در حفاظت ارگانیسم در مقابل سیستئین پروتئاز های آزاد شده ی داخلی وخارجی است.
توالی اسید آمینه ای سیستاتین c انسانی ،اولین توالی از سیستاتین برد که شناسایی شد . سیستاتین cیک پروتئین با وزن مولکولی کم به عنوان یک مهار کننده ی سیستئین پروتئاز های مختلف در خون یافت شده است.آن هر دو پروتئازهای اندوژن مثل کاتپسین های لیزوزومال و پروتئازهای پارازایت ها و میکروارگانیسم هارا مهار می کند.
سیستاتین c محتوی 120 اسید امینه است که توسط یک ژن 7.3 Kbp در کروموزوم 20 کد می شود که یک ژن housekeeping است.
موتاسیون درLeu 68 Gln در سکانس پروتئینی سیستاتین c به طور مستقیم مرتبط با گسترش آنژیوپاتی آمیلوئیدارثی سیستاتین c ( HCCAA)است که بیماران از هموراژی مغزی تکرار شونده رنج می برند.
5. اهمیت بالینی سیستاتین c
به منظور تشخیص آزمایشگاهی خیلی از بیماری ها انجام می شود:
-ارزیابی سرعت فیلتراسیون گلومرولی(GFR): غلظت یک ارتباط عالی با تصفیه ی گلومولی دارد چون تحت تأثیر سن جنس نژاد ،رژیم غذایی و...قرار نمی گیرد.
-آنوریسم واسکولار
-هایپر هموسیستئینمیا
-بیماری لنفوپرولیفراتیو
تشخیص و درمان بیماری ها
-بیماری آلزایمر
- لکوانسفالوپاتی با دمنتیا پیشرونده
-بیماری دژنراتیو شبکیه
- remodellingاستخوان
- نقص سد خونی مغزی
- آنژیوپاتی ارثی آمیلو ئید hereditary cystatin C amyloid angiopathy (HCCAA)
-افزایش سطح خارج سلولی سیستاتین A,B,C در ملانوما که مرتبط با کاهش طول عمر است که به علت تولید توسط سلول های بدخیم است.
- افزایش سیستاتین ها در بیماری های التهابی و خود ایمنی
-فعالیت های آنتی ویروس و آنتی بیوتیک سیستاتین ها مرتبط با مهار پروتئاز های اگزوژن است.رشد ویروس های پولیو و کورونا در سلول های انسانی کشت داده شده توانست با اضافه کردن سیستاتین مرغی یا سیستاتینD یا سیستاتینC به تاخیر بیفتد.
علت این که علاقه مندی به این مهارکننده های زیاد شده به خاطر این است که پروتئولیز کنترل نشده می تواند منجر به آسیب برگشت ناپذیر مثل التهاب مزمن آسیب ریه رشد بدخیمی و نیز آترو اسکلروز زودرس شود.
-سیستاتینC فعالیت ضد ویروسی دارد.
6. فاکتور های موثر بر تولید سیستاتینC
با توجه به تولید ثابت سیستاتینC فاکتور هایی که روی تولید آن اثر دارند مهم اند:
*دوزهای خیلی زیاد گلوکوکورتیکوئید ها باعث افزایش تولید سیستاتینc می شود،در حالی که دوزهای کم و متوسط گلو کو کو رتیکو ئیدها تولید سیستاتینc را تغییر نمی دهند.
*حاملگی
*اختلال عملکرد تیروئید
7.پیوند کلیه و سیستاتین c
پیوند کلیه درمان انتخابی برای مرحله ی آخر بیماری کلیه یا(End Stage Renal Disease) ESRD است ، جهت طولانی تر کردن زندگی ، بهتر شدن کیفیت زندگی و کمتر شدن هزینه ها در مقایسه با دیالیزمی باشد.اگرچه پتانسیل کامل انجام شدن این درمان محدود شده است،چون خیلی از پیوند های کلیه قبل از موعد با شکست مواجه می شوند.اگرچه بیماران بعد از شکست پیوند می توانند به دیالیز برگردند،اما از دست رفتن عملکرد پیوند مرتبط با افزایش 3برابری خطر مرگ،کاهش قابل توجه در کیفیت زندگی برای کسانی که زنده می مانند و افزایش 4 برابری در هزینه ها، است. بنابراین ارزیابی صحیح و دقیق عملکرد کلیه در بیماران بعد از پیوند کلیه ، اهمیت بالایی دارد.
اندازه گیری مستقیم GFR با استفاده از یک عامل اگزوژن دقت زیادی در برآورد عملکرد کلیه را می دهد، اما گران ، زمان بر ، طاقت فرسا و به طور وسیع قابل استفاده نمی باشد و در همه مراکز وجود ندارد. ناتوانی درتعیین درست تغییرات در GFR ،عوارض مهمی در پیوند کلیه دارد. تغییرات در GFR می تواند پیشرفت بیماری کلیه را مشخص کند ،سطح GFR ، پیش بینی قوی شروع زمان ناتوانی کلیه و خطر عوارض بیماری مزمن کلیه مثل بیماری های قلبی عروقی ،فشار خون،آنمی ،سوءتغذیه،بیماری استخوانی ،نوروپاتی و کاهش کیفیت زندگی و مرگ را می کند. کاهش تصفیه کردن پیوند ؛با کاهش بقاء گیرنده ، از دست رفتن پیوند ، مشکلات قلبی و افزایش هزینه مرتبط است.غلظت های SCr ،B2MG , BUN،و یا UA به طور معمول برای عملکرد کلیه استفاده می شوند که البته با ضعیف بودن حساسیت و صحت آن ها ،کاربرد بالینی آن ها محدود می شود.
به همین منظور،سیستاتینC (Cyc C) که یک پروتئین کوچک اندوژن است، به عنوان یک مارکر عملکرد کلیه تحقیق شده است. مزیت اصلی Cyc CبرCrاین است که فقط عملکرد کلیه را منعکس می کند. در پیوند کلیه،Cyc Cبه عنوان یک مارکر بهتر عملکرد کلیه نسبت به Crتوصیه شده است وصحت تشخیصی بالاتری دارد،چون به طور عالی قادر است ناتوانی عملکرد را منعکس کند.
8.مراجع
1. Abbud-Filho M, Adams P, Alberu J, et al. A report of the Lisbon Conference on the care of the kidney transplant recipientTransplantation. 2007;(Suppl 8)83:1-22.
2. KDOQI US commentary on the 2009 KDIGO clinical practice guideline for the care of kidney transplant recipients. Am J Kidney Dis. 2010;56:189-218.
3. Kidney Disease: Improving Global Outcomes. Transplant Work Group. KDIGO clinical practice guideline for the care of kidney transplant recipients. Am J Transplant. 2009;9(Suppl 3):S1-S157.
4. Renal Function Tests an overview !!! Dr. S.M.H.DUHS
5. Nephrol Dial Transplant (2008) 23: 154160doi: 10.1093/ndt/gfm661
6. Joanna Pollak-PRODUCTION OF CYSTATIN C WILD TYPE AND STABILIZED MUTANTS
7. G. Filler et al. / Clinical Biochemistry 38 (2005) 1–8
8. Anders Grubb, Dept. of Clinical Chemistry, University Hospital, S-22185 Lund, Sweden
9. Joanna PollakPRODUCTION OF CYSTATIN C WILD TYPE AND STABILIZED MUTANTS
10. Biomed. Papers 147(2), 177–180 (2003)© J. Mareš, D. Stejskal, J. Vavroušková, K. Urbánek, R. Herzig, P. HluštíkUSE OF CYSTATIN C DETERMINATION IN CLINICAL DIAGNOSTICS
11. Organ Transplantation 3.Diagnosis and prevention of chronic kidney allograft loss.Brian J Nankivell, Dirk R J Kuypers. www.thelancet.com Vol 378 October 15, 2011
12. C.A. White et al. / Transplantation Reviews 24 (2010) 18–27
اندازه گیری تری گلیسیرید سرم به روش آنزیمی،کالریمتری
بسمه تعالی
نام آزمایش : اندازه گیری تری گلیسیرید سرم به روش آنزیمی،کالریمتری
مقدمه:
در انسان ها اصلی ترین چربی رژیم غذایی، تری گلیسیرید است که از منابع چربی گیاهان و جانوران که بخشی از غذاهای مصرفی هستند،به دست می آید. تری گلیسیرید تا حدود 90%از راه خوراکی وارد بدن می شود.
تری گلیسیرید های غذایی که در آب نامحلول اند،در روده کوچک به اسیدهای چرب و2-منوگلیسیریدها تجزیه می شوند.این محصولات هضم شده در سلول های اپی تلیال روده ،دوباره به هم پیوسته ، تری گلیسیریدهای (اگزوژن یا برون زاد) را تولید می کنند و شیلومیکرون ها را از طریق لنف به خون ترشح می کنند(یعنی روده ، تری گلیسیرید ها را از اسیدهای چرب رژیم غذایی تولید می کند و تری گلیسیرید به صورت شیلومیکرون که قطرات ریز چربی اند و توسط پروتئین پوشیده شده اند،در جریان خون حمل می شوند). تری گلیسیرید های شیلومیکرون به وسیله ی لیپوپروتئین لیپاز ،در مویرگ های خونی تجزیه می شوند.
کبد نیز مسئول سنتز تری گلیسیرید ها است ولی این تری گلیسیرید ها به صورت لیپوپروتئین با چگالی کم(VLDL) حمل می شوند(تری گلیسیرید اندوژن یا درون زاد).
بیشتر تری گلیسیرید ها به صورت لیپید در بافت چربی ذخیره می شوند(حدود 90%چربی ذخیره بدن)،وظیفه ی تری گلیسیرید ها ،تامین انرژی برای قلب و عضلات اسکلتی است(ذخیره انرژی بدن).به علت خاصیت آبگریز بودن تری گلیسیرید ها، این ترکیب توسط سایر لیپید ها مانند فسفولیپید ، کلسترول و پروتئین به صورت لیپوپروتئین ها انتقال می یابند.بنابراین تری گلیسیرید ها از زیرواحد های ساختمانی گلیسرول و اسید های چرب ،بیشتر در کبد تولید می شوند و بعد از هر وعده غذایی تری گلیسیرید در سلول چربی ذخیره می شود و اگر آزمایش تری گلیسیرید بعد از صرف غذا انجام شود، سرم حالت شیری رنگ یا خامه ای پیدا می کند.
مقادیر طبیعی تری گلیسیرید :
normal 40-150mg/dl
151-200mg/dl مشکوک
200mg/dl (پاتولوژیک) بالا
با بیشتر شدن تری گلیسیرید از این حد، عوارضی مانند کبد چرب را ایجاد می کند که در آن تری گلیسیرید اضافی در کبد رسوب نموده و تغییرات فیبروزی ایجاد می شود که در صورت کنترل نشدن تری گلیسیرید ،منجر به سیروز کبد می شود.
موارد افزایش سطح تری گلیسیرید سرم:
1-هایپر تری گلیسیریدمی فامیلی (یک نوع استعداد ژنتیکی است که در آن تری گلیسیرید خون فرد زیاد می شود)
2-دیابت شیرین کنترل نشده
3-الکلیسم(در صورت ادامه مصرف)
4- کم کاری تیروئید(کاهش تجزیه تری گلیسیرید)
5-رژیم پر کربوهیدرات(تبدیل کربوهیدرات اضافی به تری گلیسیرید )
6- در موارد کاهش انسولین – شوک – استرس(افزایش VLDL و در نتیجه افزایش تری گلیسیرید)
1-سؤتغذیه(رژیم غذایی این بیماران کم چربی است. تری گلیسیرید بخش عمده چربی رژیم غذایی را تشکیل می دهد.)
2-سندرم سؤجذب(این بیماران دچار سؤجذب چربی های رژیم غذایی هستند، تری گلیسیرید جزء اصلی رژیم غذایی است و به دلیل جذب کم از مجاری گوارشی ، سطح تری گلیسیرید هم کاهش می یابد)
3-پرکاری تیروئید(افزایش تجزیه VLDL که لیپوپروتئن اصلی ناقل تری گلیسیرید است و در نتیجه تری گلیسیرید کاهش می یابد)
بررسی خطر ابتلا به بیماری های قلبی-عروقی آترواسکلروز
برای افراد مشکوک به اختلالات متابولیسم چربی ها و هیپرلیپیدمی
-فرد باید 12 تا 14 ساعت ناشتا باشد (فقط خوردن آب مجاز است)
-استرس نداشته باشدو 24 ساعت قبل از آزمایش نباید الکل مصرف کند.
1.خوردن غذای چرب قبل از آزمایش، باعث افزایش تری گلیسیرید می شود.
2.مصرف الکل قبل از آزمایش ،باعث افزایش تولید VLDL و افزایش تری گلیسیرید می شود.
3.بارداری باعث افزایش تری گلیسیرید می شود.
4.مصرف قرص های ضد بارداری و استروژن ها باعث افزایش تری گلیسیرید می شود.
5.مصرف استاتین ها و آسکوربیک اسید باعث کاهش تری گلیسیرید می شود.
6.تغییر وضعیت از نشسته یا ایستاده به حالت دراز کش ،میزان تری گلیسیرید را به طور چشمگیری زیاد می کند.
7.ورزش کردن باعث پایین آمدن تری گلیسیرید در عرض چند ساعت می شود.
تری گلیسیرید ها توسط آنزیم لیپاز هیدرولیز شده و اسیدهای چرب و گلیسرول آزاد می شوند،سپس طی مراحل زیر پراکسیدهیدروژن آزاد می شودکه با 4-آمینو آنتی پیرین و فنل و در مجاورت آنزیم پراکسیداز تشکیل کمپلکس رنگی کینونیمین می شود.شدت رنگ حاصل متناسب با مقدار تری گلیسیرید موجود در نمونه است.
لوله بلانک
لوله استاندارد
لوله تست
استاندارد
-
10 میکرولیتر
-
سرم
-
-
10 میکرولیتر
آب مقطر
10 میکرولیتر
-
-
معرفR1
1میلی لیتر
1میلی لیتر
1میلی لیتر
*لوله ها را مخلوط کرده ،10 دقیقه در بن ماری 37 درجه انکوبه کرده و جذب نمونه و استاندارد را در مقابل بلانک معرف در 500 نانو متر بخوانید.(پایداری رنگ 60 دقیقه دور از نور مستقیم )
تهیه و تنظیم:صالحی پور