بیوشیمی در بالین
بیوشیمیبیوشیمی در بالین
بیوشیمیدرباره من
 سلام.من سعی می کنم تست های آزمایشگاهی و کاربرد بالینی و نحوه اندازه گیری آن ها رو در این جا بذارم.امیدوارم به دردتون بخوره.
ادامه...
سلام.من سعی می کنم تست های آزمایشگاهی و کاربرد بالینی و نحوه اندازه گیری آن ها رو در این جا بذارم.امیدوارم به دردتون بخوره.
ادامه...
آمینواسیدوری
![]() مقدمه:
مقدمه:
سال های متمادی از آزمایش ادرار برای غربالگری بیماری های متابولیک استفاده شده است،به خصوص بیماری هایی که ناشی از یک استعداد ژنتیکی بوده اند.در بسیاری از این بیماری ها یک متابولیت غیرطبیعی و یا مقدار بیش از حد یک متابولیت طبیعی در ادرار دفع می شود.
به لحاظ غیر شایع بودن این بیماری ها و غیر اختصاصی بودن علائم آن ها از یک سو و قابل درمان بودن آن ها در صورت تشخیص سریع از سوی دیگر،خون و ادرار را باید با استفاده از تکنیک های بسیار انتخابی و حساس مورد آزمایش قرار داد.
![]() آمینو اسیداوری
آمینو اسیداوری
آمینو اسید در خون از میان غشای گلومرولی فیلتر می شوند ولی به طور نرمال در توبول های کلیوی توسط مکانیسم های انتقال فعال قابل اشباع بازجذب می شوند.
آستانه کلیه برای آمینو اسیدها ی پلاسما بالا است، در نتیجه در حالت طبیعی آمینو اسیدهای بسیار کمی از خون به داخل ادرار فیلتره می شوند. آمینو اسیدها یی که به وسیله ی گلومرول به داخل فیلتره ی گلومرولی وارد می شوند توسط توبول ها مجددا جذب می شوند. آمینو اسیدهایی که در حالت طبیعی به مقدار زیاد (25-200mg/24h) در ادرار دیده می شوند شامل آمینو اسیدهای گلیسین ، تورین ، هیستیدین و گلوتامین می باشند. آمینو اسیدهای دیگر نظیر تریپتوفان ،تیروزین ،سرین ،لوسین ،سیستئین ،آرژنین و فنیل آلانین به مقدار کم (0-25mg/24h) در ادرار افرادسالم وجود دارند.
دفع یک یا تعداد بیشتری از آمینو اسیدها در ادرار می تواند به علت مانعی در مسیر متابولیک (نوع سرریزی) یا کمبود در عملکرد توبولی کلیه (نوع کلیوی) باشد.
![]() تقسیم بندی آمینو اسیداوری روش دنتس
تقسیم بندی آمینو اسیداوری روش دنتس
1. آمینو اسیداوری کلیوی (Renal Aminoaciduria)
آمینو اسیداوری کلیوی معمولا در اثر ضعف(نقص) توبول های کلیوی در بازجذب آمینو اسیدها در ادرار بیمار ظاهر می شود.سطوح پلاسمایی نرمال هستند ولی یک نقص مادرزادی یا اکتسابی در سیستم انتقال کلیوی وجود دارد. آمینو اسیداوری توبولی به 2 دسته تقسیم می شود:
I. آمینو اسیداوری عمومی توبولی (Generalized Tubular Aminoaciduria)که در موارد زیر وجود دارد:در سندرم فانکونی – سیستینوزیس – بیماری ویلسون – گالاکتوزومی
II. آمینو اسیداوری اختصاصی توبولی(Specific Tubular Aminoaciduria) شامل موارد خاص است: سیستین اوری- گلیسین اوری
2. آمینو اسیداوری سر ریز (Overflow Aminoaciduria)
در این جا کلیه هیچ نقصی ندارد .به هر علتی آمینو اسیدهای پلاسما افزایش پیدا کنند که از آستانه ی کلیوی تجاوز می نمایند ، در ادرار بیمار ظاهر می شوند. آمینو اسیداوری سرریز در موارد زیر دیده می شود:
I. عمومی : بیماری های کبدی(چون محل سنتز پروتئین ها در کبد است) – نوزادان نارس (چون هنوز متابولیسم پروتئین ها خوب صورت نمی گیردو به دلیل نقص در متابولیسم پروتئین ها دفع آمینواسیدها در ادرار زیاد است )– آنمی مگالوبلاستیک – مسمومیت با سرب – تحلیل عضلانی– لوسمی
II. اختصاصی : شربت افرا(MSUD) – هارت ناپ – فنیل کتونوری
3.دفع مرضی و غیر طبیعی مشتقات آمینو اسیدها
چنانچه متابولیسم آمینو اسیدها در بدن به روال طبیعی صورت نگیرد، در اثر متابولیسم ناقص ،یک سری موادغیر طبیعی ایجاد می شوند که در ادرار بیمار دفع می شوند.در این حالت خود آمینو اسید در ادرار نیست بلکه مشتقات آن در ادرار ظاهر می شود مثل آلکاپتنوری( که آلکاپتون در ادرار ظاهر می شود)– تیروزینوز -هموسیستینوری
برای پی بردن به نوع آمینو اسید های موجود در ادرار از روش های کروماتوگرافی کاغذی، ستونی یا لایه نازک استفاده می کنند.
![]() آمینو اسید اوری ممکن است اولیه یا ثانویه باشد:
آمینو اسید اوری ممکن است اولیه یا ثانویه باشد:
1. آمینو اسید اوری اولیه
به علت یک نقص آنزیمی ارثی است که اختلال متابولیسم مادرزادی نیز نامیده می شود.نقص در مسیری که یک آمینو اسید خاص متابولیزه می شود یا نقص در سیستم انتقال توبولی خاص کلیوی واقع شده است.
2. آمینو اسیداوری ثانویه
به علت یک بیماری در یک عضو مثل کبد که یک محل فعال متابولیسم آمینو اسید است یا اختلال عملکرد توبولی کلیوی ایجاد شده یا سوءتغذیه انرژی پروتئین
![]() مقادیر طبیعی:
مقادیر طبیعی:
به طور متوسط در بالغین در طول 24 ساعت مقدار 50-200mg آمینو اسید نیتروژن از طریق ادرار دفع می شود.نوزادان به ازاء هر کیلوگرم وزن بدن مقدار 8mg نیتروژن در 24 ساعت دفع می کنند. در نوزادان نارس دفع آمینو اسید های ادرار 4 برابر نوزادان کامل می باشد.درکودکان ، دفع آمینو اسید نیتروژن در 24 ساعت برابر3mg/kg می باشد.
در سیستین اوری( آمینو اسید سیستین) –در فنیل کتونوری(فنیل آلانین)- در بیماری شربت افرا(لوسین و ایزولوسین و والین)-در بیماری هارت ناپ ( گلوتامین و هیستیدین و والین ) در ادرار دیده می شود.
![]() سندرم فانکونی:
سندرم فانکونی:
- به وسیله ی ژن اتوزومال مغلوب به افراد منتقل می شود ، در این بیماری مکانیسم جذب مجدد آمینواسیدها در لوله های ادراری دچار اختلال شده است و از نظر ظهور در فرد به دو صورت دیده می شود:
الف- سندرم فانکونی مادرزادی نوزادان
ب- سندرم فانکونی بالغین
- تشخیص آزمایشگاهی: اگرچه این دو سندرم از لحاظ بالینی با هم متفاوت اند اما از نظر تشخیص آزمایشگاهی نظیر هم می باشند.بیمار مبتلا به گلوکزاوری و پلی اوری با وزن مخصوص ثابت می باشد و دفع پتاسیم و فسفر و اسیداوریک افزایش می یابد در حالی که اسیداوریک خون کاهش می یابد.
- سندرم فانکونی نوزادان اغلب با سیستینوزیس همراه است ؛ در نوع مادرزادی، سیستین در سلول های رتیکولوم مغزاستخوان رسوب می کند که در بیوپسی اندام هایی نظیر کلیه و کبد سیستین را می توان دید.
![]() بیماری ویلسون:
بیماری ویلسون:
- یک نابسامانی متابولیکی و بسیار نادر است که در این بیماری سرولوپلاسمین پلاسما کاهش می یابد (سرولوپلاسمین پروتئینی است که در پلاسما به مس متصل می شود) . بیمار مبتلا به ضعف قوای عضلانی است و حرکاتش کند و بطئی می باشد. حلقه خاکستری مایل به سبز در اطراف چشم بیمار دیده می شود،این حلقه به نام Kayser Fleischer Ring موسوم است.
-یافته های آزمایشگاهی : پایین بودن سطح مس خون – افزایش دفع مس از طریق ادرار – آمینواسیداوری – فسفات اوری، فسفاتمی – کاهش اوریک اسید خون
![]() گالاکتوزومی:
گالاکتوزومی:
- یک بیماری ارثی که توسط ژن اتوزومال مغلوب منتقل می شود.در این بیماری گالاکتوز به گلوکز تبدیل نمی شود. ادرار بیمار حاوی گالاکتوز به مقدار زیاد است و نیز به میزان قابل توجهی دارای آمینواسید می باشد.اگر بیمار تحت درمان سریع قرار نگیرد کبد و مغز او آسیب می بینند.
![]() اختلالات متابولیسم آمینو اسیدها
اختلالات متابولیسم آمینو اسیدها
بسیاری از این بیماری ها نادر و بعید هستند که بیشتر پزشکان با آن مواجه می شوند که اگر درمان نشوند، خیلی از این اختلالات ژنتیکی باعث آسیب مغزی برگشت ناپذیر و مرگ و میر زودرس می شوند.بنابراین تشخیص قبل از تولد یا اوایل پس از تولد و شروع سریع درمان مناسب، در صورت موجود بودن لازم است.
هارت ناپ(Hurtnup)
- یک بیماری ارثی(صفت اتوزومال مغلوب نادر) است که نقص در انتقال(حمل و نقل) روده ای و کلیوی آمینواسیدها ی خنثی از جمله تریپتوفان است که بیشتر (اما نه تمام) شواهد بالینی را می توان به کاهش جذب روده ای و افزایش دفع ادراری تریپتوفان نسبت داد. در این بیماری توبول های کلیوی قادر به جذب مجدد تریپتوفان نیستند. یک آمینواسیداوری عمومی خنثی وجود دارد .
- افزایش دفع مشتقات ایندول که از تخریب تریپتوفان جذب نشده که توسط باکتری های روده به وجود می آیدحاصل می شوند ،از روده جذب شده و در ادرار دفع می شوند.
- به طورطبیعی بخشی از این آمینو اسید به نیکوتین آمید تبدیل می شود،این تبدیل به ویژه اگر مصرف خوراکی نیکوتین آمید در محدوده مرزی باشد ،اهمیت دارد.
- اختلال در جذب روده ای و بازجذب کلیوی تریپتوفان ،در دسترس بودن تریپتوفان را برای بیوسنتز نیاسین محدود می کند و باعث بروز علایم مشابه پلاگر می شود که شامل:
راش قرمز پوسته دار روی نواحی برهنه پوست
آتاکسی مخچه ای برگشت پذیر
اختلال روانی با درجات متغیر
- تشخیص : در این بیماری حداقل 10 نوع آمینو اسید در ادرار بیمار دفع می شود که با روش TLC به خوبی قابل تشخیص می باشد.
1.اختلالات متابولیکی گلیسین
a) گلیسین اوری
توسط ترشح ادراری 0.6-1 gr از گلیسین در روز مشخص می شود که یک گرایشی به تشکیل سنگ های کلیوی اگزالات دارد . گلیسین اوری احتمالاً نتیجه ای از یک نقص در بازجذب توبولی کلیوی است جایی که سطوح گلیسین پلاسمایی نرمال است.
(b هایپراگزالوری اولیه
- در این حالت، ترشح ادراری اگزالات غیر وابسته به جذب رژیم غذایی اگزالات است.
- اگزالات ظاهراً از دآمینه شدن گلیسین افزایش می یابد،با تشکیلGlyoxylate (اگزالات سمی آلدهید)
- نقص متابولیکی شامل یک نارسایی برای کاتابولیزه شدن Glyoxylate است که از این رو به اگزالات اکسید می شود:
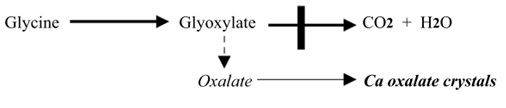
- خصوصیات بالینی شامل سنگ اگزالات کلسیم دو طرفه –عفونت مکرر مجاری ادراری است که با مرگ زودرس از نارسایی کلیوی یا فشار خون دنبال می شود.
- درمان : مصرف پیریدوکسال فسفات (ویتامینB6 ) که یک کو ترانس آمیناز است که ترانس آمیناسیون Glyoxylate را دوباره به گلیسین تحریک می کند.
2.اختلالات متابولیکی آمینواسید های محتوی سولفور
(a Cystinuria (Cystine-Lysinuria)
- یک بیماری متابولیکی ارثی و اتوزومال مغلوب است که با ترشح ادراری سیستین بالای 30 برابر نرمال مشخص می شود که ترشح لیزین ، آرژنین و اورنیتین نیز افزایش می یابد.
- نقص در مکانیسم های بازجذب کلیوی توسط توبول های کلیوی برای این 4 آمینواسید است و وجود سیستین، لیزین ، آرژنین و اورنیتین در ادرار دلیل بر سیستین اوری است و با سیستینوزیس متفاوت است. در این بیماری سطح این 4 آمینواسید در خون بیمار طبیعی یا پایین تر از طبیعی است.
- یک نقص انتقالی مشابه در مخاط روده نشان داده شده است ، اما با وجود کاهش آمینواسیدهای دی بازیک ، به دلیل امکان سنتز آن ها در بدن ، کمبود های آن ها ایجاد نمی شوند.
- از آن جا که سیستین نسبتا نامحلول است،تشکیل سنگ سیستینی در توبول های کلیوی بیماران سیستینوریک رخ می دهد که این نشانه در سیستینوزیس دیده نمی شود.این حالت در افرادهموزیگوت وجود دارد یعنی امکان رسوب و ساختن سنگ در مجرای کلیه وجو دداردولی در هتروزیگوت ها افزایش دفع را می توان نشان داد اما به ندرت در حدی است که ایجاد رسوب کند.
- تشخیص آزمایشگاهی : با روش TLC به راحتی می توان به وجود این 4 آمینواسید در ادرار بیمار پی برد.
- درمان : کاهش دادن غلظت سیستین در ادرار با نوشیدن مقادیر زیادی آب است .قلیایی کردن ادرار، حلالیت سیستین را بالا می برد.اگر این اقدامات ناکافی باشند ، می توان D – پنی سیلامین تجویز کرد ،این دارو با سیستین کمپلکسی می سازد که بسیار محلول تر از سیستین به تنهایی است.
(b سیستینوزیس(بیماری ذخیره سیستین)
- یک اختلال متابولیکی ارثی(اختلال لیزوزومال نادر) است که این وضعیت باید از سیستینوری که یک وضعیت نسبتاً بی خطر است افتراق داده شود.
- نقص در Carrier-mediated Transporter سیستین است.
- کریستال های سیستین در بافت ها و اندام ها خصوصا سیستم رتیکولواندوتلیال رسوب می کنند.با رسوب داخل سلولی کریستال سیستین درون لیزوزوم ها مشخص می شود.کریستال ها ممکن است در کلیه – چشم- مغز استخوان و طحال تجمع کنند.
- در شکل شدید این بیماری،فتوفوبی،نارسایی کلیه ، ریکتز و اختلال رشد دیده می شود.
- با درگیری و آسیب توبول های کلیه به وسیله ی سیستین ، باعث ایجاد سندرم فانکونی می شود و گلوکزاوری و آمینواسیداوری عمومی حادث می شود.
- برخلاف سیستینوری،از دست رفتن سیستین در سیستینوزیس به موازات از دست رفتن سایر آمینواسیدها در ادرار دیده می شود و سیستینوزیز معمولا با آمینواسیداوری عمومی و با منشا کلیوی همراه است. این حالت در سندرم فانکونی دیده می شود که نباید با سیستین اوری اشتباه شود.
- سایر عملکرد های کلیوی نیز به شدت دچار نقص می شوندو بیماران معمولا در جوانی از نارسایی حاد کلیوی می میرند.
(c هموسیستینوریا
- نقص ارثی کاتابولیسم متیونین که توسط ژن مغلوب منتقل می شود.
- در این بیماری متابولیسم متیونین دچار اختلال شده و مقدار زیادی هموسیستین در ادرار بیمار وجود دارد.
نقص به خاطر کمبود سیستاتیونین β سنتاز(CS) است ، ماده ای که تشکیل سیستاتیونین را از هموسیستین و سرین در مسیر متیونین کاتالیز می کند.
- بالای 300میلی گرم هموسیستین ، گاهی با هم با S-آدنوزیل متیونین روزانه در ادرار ترشح می شوند.سطوح پلاسمایی متیونین نیز افزایش می یابد.
- یافته های بالینی شامل عقب ماندگی ذهنی– تشنج-انگشتان عنکبوتی(arachnodactuly)-پوکی استخوان-ترومبوز-تغییرات استخوانی عروقی و تغییراتی که در عدسی چشم بیمار دیده می شود است.
- نمونه ادرار باید تازه باشد زیرا هموسیستین ناپایدار است.
- سیستینوری و هموسیستینوری را می توان با استفاده از روش الکتروفورز با ولتاژ قوی از یکدیگر تشخیص داد.
- درمان : با کاهش متیونین در رژیم غذایی و تجویز پیریدوکسین است.
3.اختلالات متابولیکی فنیل آلانین و تیروزین
(a فنیل کتونوری (PKU)یا فنیل آلانینمیا
- یک بیماری ارثی اتوزومال مغلوب است.اختلالات عمده متابولیکی در ارتباط با اختلال توانایی برای تبدیل فنیل آلانین به تیروزین است.
- فنیل آلانینمی : فنیل آلانین یکی از آمینواسیدهای ضروری بدن می باشد که از متابولیسم این آمینواسید مواد مختلفی تولید می شود. فنیل آلانینمی زمانی اتفاق می افتد که در یکی از آنزیم هایی که در متابولیسم این آمینواسید دخالت دارند کمبود یا فقدان وجود داشته باشد.فنیل آلانینمی با PKU همراه است.

- نقص در این مسیر تبدیلی ممکن است به علل زیر باشد:
1.فقدان یا کمبود فنیل آلانین هیدروکسیلاز(PKU کلاسیک یا نوع I)
2.نقص در دی هیدروبیوپترین ردوکتاز (نوع II و III)
3. نقص در دی هیدروبیوپترین بیوسنتاز(نوع IV وV )
- مهم ترین پیامد PKU نوع I درمان نشده ، عقب ماندگی ذهنی،تشنج ، اگزما و بوی موش است.
- سطوح پلاسمایی فنیل آلانین و فنیل پیروییک اسید و سطح ادراری فنیل پیروییک اسید(بالاترین) ، فنیل استیک اسید و فنیل آلانین افزایش نشان می دهند.
- یافته های آزمایشگاهی شامل : افزایش فنیل آلانین و متابولیت های فنیل آلانین (فنیل پیروییک اسید- فنیل لاکتیک اسید – فنیل استیک اسید ) در خون و ادرار است.
- به عنوان علامت مشخصه ادرار و عرق این بیماران به علت فنیل استیک اسید، دارای بوی موش یا کپک می باشد.
-تیروزین خون بعد از Loud فنیل الانین افزایش نمی یابد.
- درمان : با محدودیت فنیل آلانین در رژیم غذایی

تیروزینمیا(تیروزینوز)-تیروزینوری
- یک نارسایی متابولیسمی و یک بیماری ارثی است.
- تیروزینمی همراه با تیروزینوری زمانی اتفاق می افتد که تیروزین مشتق از رژیم غذایی یا فنیل آلانین دارای متابولیسم غیر طبیعی باشد که ممکن است قسمتی از یک اختلال عمومی آمینواسیدها همراه با بیماری کبدی یا نشان دهنده ی یکی از اختلالات ژنتیکی متابولیسم تیروزین باشد.
- نقص : تیروزینمی ارثی نوعI(تیروزینوز) : کمبود فوماریل استواستات هیدرولاز(FAA Hydrolase)
تیروزینمی نوعII : کمبود تیروزین ترانس آمیناز
- یافته های آزمایشگاهی شامل : افزایش تیروزین و متابولیت های آن پارا- هیدروکسی فنیل پیروییک اسید ،استیک و لاکتیک اسیدها
- یافته های بالینی : سیروز کبدی،انبساط شکمی ،بزرگی طحال ، آسیب کلیوی که ممکن است باعث مسمومیت آمونیا(ammonia) و باعث گلوکزاوری کلیوی شود.
- درمان : محدود کردن رژیم غذایی فنیل آلانین – تیروزین –متیونین
1.فقدان یا کمبود فنیل آلانین هیدروکسیلاز(PKU کلاسیک یا نوع I)
2.نقص در دی هیدروبیوپترین ردوکتاز (نوع II و III)
3. نقص در دی هیدروبیوپترین بیوسنتاز(نوع IV وV )
- مهم ترین پیامد PKU نوع I درمان نشده ، عقب ماندگی ذهنی،تشنج ، اگزما و بوی موش است.
- سطوح پلاسمایی فنیل آلانین و فنیل پیروییک اسید و سطح ادراری فنیل پیروییک اسید(بالاترین) ، فنیل استیک اسید و فنیل آلانین افزایش نشان می دهند.
- یافته های آزمایشگاهی شامل : افزایش فنیل آلانین و متابولیت های فنیل آلانین (فنیل پیروییک اسید- فنیل لاکتیک اسید – فنیل استیک اسید ) در خون و ادرار است.
- به عنوان علامت مشخصه ادرار و عرق این بیماران به علت فنیل استیک اسید، دارای بوی موش یا کپک می باشد.
-تیروزین خون بعد از Loud فنیل الانین افزایش نمی یابد.
- درمان : با محدودیت فنیل آلانین در رژیم غذایی

آلکاپتنوری
- یک اختلال متابولیکی ارثی است که توسط ژن مغلوب به ارث می رسد.
- به طور طبیعی فنیل آلانین و تیروزین به هموژنتیسیک اسید (دی هیدروکسی فنیل استیک اسید) متابولیزه می شوند، که آن هم به مالئیل استو استیک اسید اکسید می شود.
- نقص به دلیل فقدان هموژنتیسیک اسید اکسیداز کبدی است که افزایش هموژنتیسیک اسید که به طور طبیعی از متابولیسم فنیل آلانین ایجاد می شود که دربافت ها و خون تجمع می یابد، وجود دارد که در ادرار می ریزد.
- اکسیداسیون و پلی مریزاسیون هموژنتیسیک اسید باعث تولید پیگمان آلکاپتون می شود که مسیر آن شبیه پلی مریزاسیون DOPA برای تولید ملانین است.
- بیمار در سنین جوانی ناراحتی به خصوصی ندارد ولی با بالا رفتن سن ، پیگمان های سیاهی در استخوان و مفاصل ظاهر شده و بیماری خاصی به نام اکرونوزیس به وجود می آید که در اثر رسوب آلکاپتون در غضروف ها و تیرگی حاصل از ان است و سبب تیرگی غضروف های گوش و معمولاً آرتریت در سنین بالا می شود.
- یافته های بالینی : آرتریت(التهاب مفاصل) دژنراتیو – پیگمانتاسیون غضروف
- تشخیص : با آزمایش هموژنتیسیک اسید در ادرار- روش های تاییدی شامل کروماتوگرافی کاغذی یا لایه نازک و الکتروفورز موئینگی است.این روش ها باید هموژنتیسیک اسید را از ژانتیزیک اسید ( یک متابولیت آسپرین) افتراق دهند.
- ادراری که حاوی این اسید باشد ظاهری معمولی دارد ولی در اثر ماندن یا قلیایی شدن به رنگ قهوه ای یا سیاه در می آید.تبدیل هموژنتیسیک اسید به آلکاپتون در شرایط قلیایی تسریع می شود و مشخص ترین ناهنجاری در آلکاپتنوری ،تیرگی ادرار است که با راکد ماندن قلیایی تر شده است،اما این یافته همیشه وجود ندارد.در صورت وجود ، اولین بار مادر بچه از این وضعیت نگران می شود به این صورت که کهنه ی بچه سیاه است و با شستشو در شوینده های قلیایی سیاه تر می شود.
- درمان : در دسترس نیست.
4.اختلالات متابولیکی متابولیسم آمینواسیدهای شاخه دار(والین – لوسین – ایزولوسین)
بیماری شربت افرا MSUD (Maple Syrup Urine Disease): Branched Chain Ketonuria
- یک اختلال مغلوب اتوزومال ارثی است که مرتبط با متابولیسم غیرطبیعی زنجیره جانبی آمینواسیدها است.
- نقص بیوشیمیایی فقدان یا کاهش شدید فعالیت α-کتواسید دکربوکسیلاز است.

- در این بیماری آمینواسید های شاخه دار مانند والین – لوسین – ایزولوسین متابولیزه نمی شوند و این آمینواسیدها به صورت α-کتواسید مربوطه اشان در خون تجمع یافته و در ادرار بیمار ظاهر می شوند.
- سطوح پلاسمایی و ادراری والین – لوسین – ایزولوسین و α-کتواسیدها افزایش می یابد.
- به علت شباهت بوی ادرار با بوی شربت افرا این نام را برای بیماری به کار می برند یا قند سوخته یا مواد قندی حامل کارامل یا زردچوبه هندی را دارد.در هر صورت علت آن نامشخص است.
- بچه به سختی غذا می خورد و ممکن است استفراغ کند و نیز تشنج ، گیجی ، تنفس های نامنظم و اغلب هیپوگلیسمی مشخص می شود.
- آسیب مغزی شدید در کودکان بازمانده رخ می دهد.بدون درمان مرگ معمولا تا پایان یک سال رخ می دهد.
- تشخیص قبل از یک هفتگی فقط توسط آنالیز آنزیمی یا ژنتیکی صورت گیردو این بیماری در هفته اول زندگی ظاهر می شودو اگر درمان صورت نگیرد ، ضایعات نورولوژیک شدید ایجاد شده و سبب مرگ در چند هفته یا ماه اول خواهد شد.
- تشخیص : نشان دادن افزایش غلظت آمینو اسیدهای منشعب در پلاسما و ادرار
- درمان در هفته اول زندگی تا حد زیادی از عواقب جلوگیری می کند.
درمان : جایگزینی پروتئین غذایی با یک مخلوطی از امینو اسید ها که شامل مقادیر اندکی از آمینواسید های با زنجیره منشعب والین – لوسین – ایزولوسین است ،تکامل نرمال محتمل خواهد بود.
سلام استاد . خوب هستید ؟ به پاس زحمات شما دوست دارم یک قالب برای وبلاگ شما طراحی کنم اگر مایلید به من ایمیل بزنید
لطفا به وبلاگ من سر بزنید
سلام آقای باوی . ممنونم .
ببخشید دیر جواب دادم.واقعا ممنون . شما لطف دارید . اتفاقا طراحی قالب رو دادم برام انجام بدن . بازم ممنون از پیشنهادتون
حتما
سلام
با تشکر از مطلب مفیدتون
چنانچه امکانش هست تست تاییدی تشخیص تیروزین در ادرار را هم بفرمایید
سلام
خواهش می کنم
در وب لاگ قرار دادم .